Gaussian09量子化學計算方法與基底函數簡介
標題 :
計算方法
HF
MPn
CI
CC
DFT
基底函數
CGTF
DZ
Split valence
Polarized
Diffuse functions
cc basis set
量子化學計算方法
| 基礎概念 |
所屬方法 |
Ab Initio Theory
(Latin,"from the beginning") |
HF,AM1,PM3,MP2,QCISD(T),CCSD(T),BD(T)
Gaussian-nCBS,Wn
CIS,TD-DFT,EOM-CCSD,CCn
CASSCF,CASPT2,MRCI,MRCI-PT2 |
| Semiempirical |
ZINDO,CNDO,INDO/S,MNDO,AM1,PM3,PM6 |
Density Functional Theory
(DFT) |
LSDA,GGA,Hybrid GGA,Double-Hybrid-Meta GGA |
| Hybrid Methods |
MCG3-DFT,MLSE-DFT |
以下介紹幾種常見方法
Hartree-Fock(HF) Method
Møller-Plesset Perturbation(MPn)
Theory
Configuration Interactions(CI)
Coupled-Cluster Method(CC)
Density Functional Theory(DFT)
Hartree-Fock(HF) Method
SCF方法(self-consistent
field)
解出 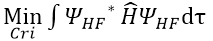 的過程(無法直接求解)
的過程(無法直接求解)
(Cri :
第i個MO中第r個基底函數的展開係數)
電腦程式產生一組近似的分子軌域(initial
guess)
 HF計算
:
得到MO數目=使用基底函數數目(K)
HF計算
:
得到MO數目=使用基底函數數目(K)
Unoccupied MOs(virtual orbitals)數目>>occupied
MOs
能量零點:
所有電子與原子核分離治無窮遠&靜止狀態
1 hartree = 627.5095 kcal/mol
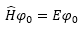
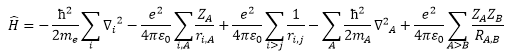
根據non-relativistic
量化理論(沒有考慮相對論效應)
無法處理spin-orbital
coupling,高速運動時,電子質量增加
只是求解近似方式,滿足close-shell分子波函數anti-symmetric(反對稱),但只有在單電子系統正確
(因為僅考慮電子平均作用力,忽略瞬間作用力)
CE(correlation energy) : HF方法未考慮的效應之能量貢獻
*現代量化計算
: HF +
不同程度近似(估計CE)
Møller-Plesset Perturbation(MPn)
Theory
根據perturbation
theory(微擾理論)
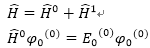
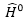 :
unperturbed
:
unperturbed
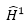 :
perturbation
:
perturbation
 :
zeroth-order基態波函數
:
zeroth-order基態波函數
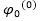 :
zeroth-order
基態能量
:
zeroth-order
基態能量
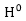 :
單電子Fock
operator之和
:
單電子Fock
operator之和
基態能量
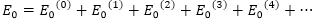
一級校正
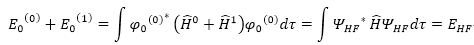
二級校正
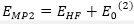
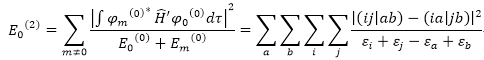
 :
有使用virtual
orbitals的較高能量之slater
determinants
:
有使用virtual
orbitals的較高能量之slater
determinants
i.j :
不同的occupied
spin orbitals
a.b :
不同的virtual
spin orbitals
MP2 :
使用兩個virtual
orbitals的
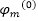
缺點
: MP2
計算量
正比
系統五次方
MP4
計算量
正比
系統六~七次方
較大系統不好用,非限制性開殼層體系不好處理(HF系列同)
Configuration Interactions(CI)
HF計算中,得到K個MO,其中n個MO可得到HF
wavefunction
將2n個電子指定於最低能階的n個軌域(configuration)
將波函數寫成所有configurations對應slater
determinants的線性組合
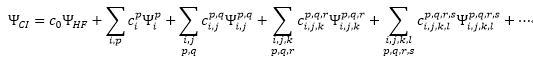
i,j,k,...: HF理論中填滿的軌域(occupied
orbitals)
p,q,r,...: HF理論中空軌域(virtual
orbitals)
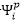 :
將一個原來在第i個填滿軌域的電子放到第p個空軌域
:
將一個原來在第i個填滿軌域的電子放到第p個空軌域
(singly excited slater determinants)
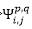 :
將第i,j填滿軌域的兩個電子放到第p,q兩個空軌域
:
將第i,j填滿軌域的兩個電子放到第p,q兩個空軌域
(doubly excited slater determinants)
係數由variational
principle (變分原理)
將能量最小化來決定
(式子包含所有可能組態,
FCI,
Full Configuraiton Interaction)
缺點 :
產生的configuration數量過於龐大
limited-CI(truncated
CI)
如CI-SD(只使用前三項),很少使用於化學反應系統,相對能量不可靠
quadratic
CI ( QCISD,QCISD(T) )
計算激發態
CI
Singles
(CIS)
式子第3項微擾
CIS(D)
Coupled-Cluster Method(CC)
基本方程式 :
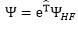 得到完整Schrödinger
equation
得到完整Schrödinger
equation
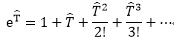
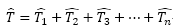
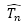 :
HF波函數產生所有n-electron
excitation之slater
determinants運算元
:
HF波函數產生所有n-electron
excitation之slater
determinants運算元
例 :
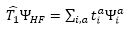
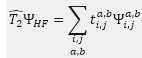
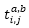 :
滿足式子,經由複雜推演導出的非線性方程式中反覆求解
:
滿足式子,經由複雜推演導出的非線性方程式中反覆求解
缺點 :
計算量太龐大
只用T2
CCD
保留T1.T2
CCSD
利用微擾理論估計T3的貢獻
CCSD(T)
QCISD(T).CCSD(T)
可靠,但須搭配適當的基底函數(如aug-cc-pVTZ)才能完全發揮功能
Density Functional Theory(DFT)
假設一個虛體系統,電子數目與實際系統相同,但電子之間並沒有作用力,每個電子都感受到一種位能(external
potential)
在這種external
potential下,虛擬系統的電子密度函數=真實系統
近似的電子密度&能量(電子波函數是一個slater
determinant)
orbital滿足
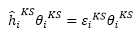
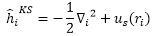
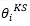 :
density orbital (Kohn-Sham orbital)
:
density orbital (Kohn-Sham orbital)
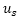 :
external potential
:
external potential
系統電子密度
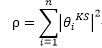
虛擬系統中電子動能
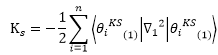
電子與原子核間位能
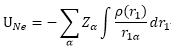
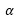 :
不同原子核
:
不同原子核
 :
核電荷
:
核電荷
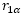 :
空間中任一點到原子核
:
空間中任一點到原子核
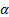 的距離
的距離
電子之間的古典靜電位能
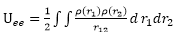 (
(
 :
避免重複計算電荷間的作用力)
:
避免重複計算電荷間的作用力)
由於與真實系統的動能有所不同,也不包含量子化學中電子間的exchange及correlation
energy
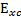 (exchange-correlation
energy functional)
(exchange-correlation
energy functional)
系統總能量
:
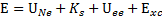
Hohenberg-Kohn variational theorem :
如果我們知道真正的density
functional,則真正的電子密度會最小化系統的能量
藉由改變電子密度分佈函數將E最小化,求得系統的電子密度.能量
因為電子密度可由KS
orbital求出
所以改變KS
orbital
影響電子密度分佈函數
最佳化以得到能量
產生類似Hartree-Fock
Equation (Kohn-Sham Equation)
可用SCF方法求解(KS
orbital)
求構成KS
orbital
最佳的基底函數的線性組合
DFT沒有包含任何近似,但因為不知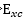 真正的數學型態
真正的數學型態
各種DFT方法差異於使用不同exchange-correlation
energy functional
只與密度函數本身有關>
Local (Spin) Density Approximation (LDA or LSDA)
與密度函數的gradient有關>
generalized-gradient qpproximation (GGA)
命名 :
以exchange及correlation
function命名
BLYP
Hybrid DFT :
在exchange中混入一定比例,類HF的exchange
energy,如B3LYP
基底函數(Basis
Function)
分子軌域(MO)
:
原子軌域的線性組合(MO-LCAO)
基底函數 :
展開分子軌域的原子軌域(AO),
所有基底函數的集合稱為基底函數組 ( basis set )
Slater-type
orbital (STO) :
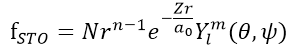
N : normalization constant
Z(zeta) : orbital exponent
傳統上用來做多電子原子及雙原子分子計算的基底函數
為類氫原子軌域的簡化,描述在原子核附近波函數行為
HF中,每一個MO是以數個STO線性組合而成
缺點:多原子分子的計算會沒有效率
Gaussian-type function(GTO) :
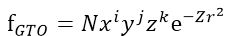
N : normalization constant
Z(zeta) : orbital exponent
i,j,k : 0 or
正整數
i+j+k =
0 s-type GTO(1種)
=
1 p-type GTO(3種)
=
2 d-type GTO(6種)
3d
AO+S
也稱Cartesian
Gaussian
AO在角度上以x.y.z取代spherical
harmonic函數
exponential項使用r平方,而非STO的r
與STO都以原子核為中心的AO
可大幅簡化雙電子積分計算
(以兩個不同原子為中心的GTO乘積
等於另一個以這兩個原子之間的點為中心GTO)
contracted
Gaussian-type function(CGTF) :
以數個GTO做線性組合成類STO函數
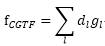
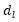 :
展開係數
:
展開係數
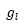 :
primitive Gaussians
:
primitive Gaussians
以同一個原子為中心的數個Cartesian
Gaussian,
但有不同的exponents(Z)
用來描述一種原子的所有CGTF稱為基底函數(basis set)
CGTF數目=同週期原子可用之原子軌域數
minimal basis set
舉例 :
碳(
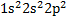 )
)
Minimal basis set :
1 s-type CGTF
1s orbital
1 s-type CGTF
2s orbital
3 p-type CGTF
2p orbital
STO-3G
basis set
每個CGTF是用3個GTO線性組合來模擬一個STO
AO
計算結果最好情況僅做到定性的預測,需增加準確度
增加基底函數的量
Double-zeta
(DZ) basis set :
對每個可用的原子軌域使用兩個CGFT描述
(計算上用到的基底函數數目也加倍)
缺點
:
計算量大增
改進 :
價軌域
DZ
內層軌域
minimal basis set
(內層軌域貢獻在計算相對能量會抵消)
split-valence double-zeta basis set
如D95(DZ)
D95V
Pople-type
basis type
Split valence basis sets (分裂價層基底函數組)
在分裂價層基底函數組(Split
valence basis set)中,
構成原子軌域的基底函數會改變大小而不會改變其形狀是它最大的特色。
例如:3-21G及6-31G就是常用的基底函數組型態。
3-21G
3 :
每一個內層電子(core
electron) orbital
是用3個primitive
Gaussians所組成一個CGTF來代表
21 :
每一個價電子軌域是由兩個CGTF代表
2 :
其中一個CGTF,
由2個primitive
Gaussian組成
1 :
另一個CGTF,
1個exponent絕對值最小的uncontracted
GTO
6-311G
6 :
每一個內層電子(core
electron) orbital
是用6個primitive
Gaussians所組成一個CGTF來代表
311 :
每一個價電子軌域是由三個CGTF代表
3 :
其中一個CGTF,
由3個primitive
Gaussian組成
11 :
2個exponent絕對值最小的uncontracted
GTO
Polarized basis sets (極化性基底函數組,表示法中有"*"的符號)
對每個原子軌域的描述中,加入較其在基態(Ground
state)所需要,
含有較高角動量(angular
momentum)的軌域型態。
(基底函數加上polarization
functions)
6-31G* or
6-31G(d) :
對第二週期(Li-Ne)及第三週期(Na-Ar)原子
加上一組uncontracted
d-type GTO
目的 :
分子的計算中較容易將電子的密度朝鍵結的方向極化,
得到較可靠的結構與能量
6-31G** or
6-31G(d,p) :
對第二週期(Li-Ne)及第三週期(Na-Ar)原子
加上一組uncontracted
d-type GTO,
對第一週期(H-He)原子加上一組uncontracted
p-type GTO
6-311G(2df,2pd) :
對第二週期(Li-Ne)及第三週期(Na-Ar)原子
加上兩組d-type
GTO與一組f-type
GTO,
對第一週期(H-He)原子加上兩組p-type
GTO與一組d-type
GTO
Diffuse functions (擴散函數,表示法中有"+"的符號)
與標準的價層軌域函數的大小相比而言,為一個擁有s軌域型態或p軌域型態的放大函數。
因此,加入擴散函數之後,軌域會擁有更大的空間,可以供給電子佔據。
含有擴散函數的基底函數組,對於電子離原子核較遠的系統非常的重要。
這些系統包括:
含有不成對電子的分子系統,
陰離子系統以及含有特定負電荷的分子系統,
如激發態系統,含低游離能的系統等等。
6-31+G* or 6-31+G** :
在6-31+G*
or 6-31+G** basis set
加入一組 s-
及一組
p-type diffuse functions
6-31++G* or
6-31++G** :
在6-31+G*
or 6-31+G** basis set
加入一組 s-
及一組
p-type diffuse functions以外,
對H與He也加入一級
s-type diffuse functions
(第一週期的效用並不明顯)
Correlation-consistent (cc) basis set (cc-pVnZ,n=D,T,Q,5,6)
著重於高階correlation
energy的計算與外插至complete
basis set (CBS) limit收斂情形
p :
polarization function,內含的
VDZ :
valence double zeta
第二週期與其之後原子的DZ中含有
d polarization functions
VTZ :
valence triple zeta
第二週期與其之後原子的TZ中含有d.f
polarization functions
aug(augmented) : aug-cc-pVDZ
第一週期含有一組
p polarization functions以外,
再加入一組s,p,d
diffuse functions
第二週期含有一組
d polarization functions以外,
再加入一組s,p,d
diffuse functions
搭配高階理論時有較好的效果
| |
H-He |
Li-Ne |
Na-Ar |
|
cc-pVDZ |
2s1p(5 function) |
3s2p1d(14 function) |
4s3p1d (18 function) |
|
cc-pVTZ |
3s2p1d(14 function) |
4s3p2d1f(30 function) |
5s4p2d1f(34
function) |
|
cc-pVQZ |
4s3p2d1f(30 function) |
5s4p3d2f1g(55 function) |
6s5p3d2f1g(59
function) |
